Chandrasekhara Venkata Raman (prix
Nobel 1930) et K.S Krishnan passent
pour être les premiers expérimentateurs de l’effet Raman lors de l’observation
de la réflexion d'un rayon de lumière solaire sur une surface d'eau
(Raman & Krishnan, 1928), mais c’est L. Brillouin et A. Smekal
qui, en 1923, firent les premières prévisions de la réflexion inélastique
de la lumière.
Ces premières observations ont
encore pendant quelques années été étayées par les travaux de Cabanes
(1928), Landsberg & Mandelstam (1928), Rocard (1928), Raman & Krishnan
(1929), et enfin ceux de Placzek (1934) avant que la théorie de « l’effet
Raman » ne sombre dans un sommeil latent …
Ce ne fut que grâce au développement
du laser (Schawlow & Townes, 1958, Mainman, 1960) que le principe Raman
renaquit ayant ainsi acquis une source d'excitation idéale (très faible
divergence, intensité élevée, monochromaticité, polarisation bien définie,
etc.). Jusqu’alors, la source de lumière excitatrice exigeait
des lampes très puissantes, le plus souvent au mercure munies de filtres
isolant l’une des raies émises (Colthup et
al. 1964 ; Encycl.Univ., 1995).
D’autres avancées technologiques
participèrent au renouveau de la Spectroscopie Raman dans les années 1950-60 :
les détecteurs à plaques photographiques, les spectrographes à prismes,
les photomultiplicateurs (cellules photoélectriques à multiplicateurs
d’électrons par émission secondaire), les filtres holographiques à haute
résolution, les détecteurs CCD, ou encore les micro-ordinateurs...
Peu de temps après, Delahaye & Migeon
(1966), Delahaye & Dhamelincourt (1975) puis Dhamelincourt et Bisson
(1977) mirent au point le principe de la MICROscopie Raman en réduisant
considérablement le point d'impact, inférieur au micromètre indépendamment de
l'intensité du signal Raman.
De nos jours, outre les applications
médicales (ex : Kline & Treado, 1997; Shim & Wilson, 1997;
Wentrup-Byrne et al., 1997), biochimiques (ex : Wynn-Williams & Edwards,
1999 ; Newton et al., 1999), extra-terrestres (ex : Hill et
al., 1999 ; Edwards et al., 1999) ou industrielles (ex :
de-Wall et al., 2000 ; Agnely et al., 2000 ;
ISA, 1984), la MR trouve de plus en plus de nouvelles applications fructueuses
dans l'analyse physico-chimique non-destructive de géomatériaux
(Smith, 1987) ou biomatériaux provenant d'objets de valeur d'intérêt artistique
ou archéologique. On peut citer les bijoux, les roches, les céramiques,
les vitraux, les métaux corrodés, les résines, les tissus (ex : Pinet et
al., 1992; Colomban, 2001 ; Colomban & Trepoz, 2001 ;
Colomban et al., 2001 ; Brody et
al., 1998 ; Fry et al., 1998 ; Smith & Edwards, 1998;
Bouchard & Smith, 2000c et Smith & Bouchard, 2000 -issus du
présent mémoire-), ou encore les pigments, qu'ils soient inorganiques
(ex : Delhaye et al., 1985; Guineau, 1987, 1991; Best et
al., 1992, Bell et al., 1997; Bussotti et al., 1995;
Rull & Alvarez, 1998; Smith & Barbet, 1999; Withnall, 1998, 1999)
ou organiques (ex : Coupry et al., 2000) qu'ils soient préhistoriques
(Bouchard, 1998; Smith et al., 1999a -issus du présent mémoire-;
Edwards et al., 1999a), historiques (ex : Vandenabeele & Moens,
1999 ; Smith & Barbet, 1999; Smith, 2000) ou contemporaines (ex :
Vandenabeele et al., 2001). Il existe par exemple un domaine bien
marqué de la MR qui est réservé aux gemmes (ex : Dhamelincourt & Schubnel,
1977; Dele et al., 1978 ; Pinet et al., 1992) : la MR
permet d'identifier les origines naturelles (géologique aussi bien que
géographique) ou artificielles des gemmes grâces aux micro-inclusions et
devient ainsi une technique de prédilèction pour l’étude de ce genre de
matériaux.
Les avantages de la MR sont nombreux, mais cette technique présente aussi
quelques limites :
Avantages :
Cette méthode
est tout d’abord non destructive -elle conserve l’objet ou l’œuvre étudié-.
On peut même faire des analyses in situ; elle ne nécessite a priori
aucune préparation avant analyse (polissage, pastillage, montage, etc.),
elle est donc idéale pour l’étude de tout objet de valeur, tels que les
objets archéologiques.
Elle ne nécessite
qu’un infime volume d’échantillon. Contrairement à la spectrométrie
Infra-Rouge (IR) qui demande quelques dizaines de milligrammes, la MR ne
nécessite qu’un volume de quelques microns cube ou même un seul microcristal.
Elle permet
l’étude de milieux non cristallisés tels que le verre, les liquides,
les gaz ou toute autre matière organique (ex : charbon de bois, résine).
Elle trouve
une application dans le domaine des Arts : il s’agit de la capacité à identifier
un produit à travers une vitre ou une quelconque protection transparente
(Smith & Rondeau, 2000). Ceci permet d'éviter de manipuler l'objet à étudier
ou de le sortir de sa vitrine ou de sa protection (ex : pour les peintures
ou les pastels). Un objectif « longue focale » est alors nécessaire
pour effectuer l'analyse.
Autre avantage
-et non des moindres-, la MR permet d'identifier les différentes sortes
de polymorphes d’une même espèce minéralogique (ex : l’aragonite et
la calcite, le quartz et la cristobalite, la goethite et
la lépidocrocite). Elle devient alors un outil complémentaire essentiel
pour toute étude optique par microscope. Cet avantage est d'autant plus
important que la nature de certains polymorphes renseigne précieusement
sur les conditions thermodynamiques de leurs formations (température,
pression, etc.). On peut citer l’exemple soulevé par G
. Turrell
(1996) concernant le minéral de formule Al
2SiO
5 dont
il est plutôt aisé de distinguer et de reconnaître les trois espèces
polymorphes (
sillimanite,
andalusite et
disthène)
par une étude
optique (dans certains cas la Spectrométrie Raman reste nécessaire),
alors que dans le cas de TiO
2, il serait plus difficile de
distinguer les différents polymorphes (
brookite,
rutile et
anatase)
uniquement par la méthode optique, leurs spectres Raman étant par contre
très différents. Notons également que, à l’instar de la DX et à l'inverse à l’analyse élémentaire,
la MR permet d’identifier
simultanément la
structure physique du
matériau analysé ainsi que sa
composition chimique élémentaire.
Enfin, la miniaturisation des instruments
est aussi un avantage majeur qui permet d’effectuer des analyses in situ grâce à des
sondes portables ou transportables (voir ce chapitre, II.2).
Limites :
Plus le minéral
est coloré ou de structure complexe, plus l’acquisition est longue -quelquefois
plusieurs dizaines de minutes-. Ceci est dû à une très forte absorption
des rayons laser par les minéraux opaques. Aussi, est-il quelquefois conseillé d’utiliser
un rayonnement laser de couleur proche de celle de l’échantillon, afin
de réduire l’absorption; ex : un laser vert à ion argon pour les cristaux
verts et un laser rouge à néon pour les cristaux rouges (cette « astuce » permet également
d’augmenter l’intensité du signal Raman par un effet de résonance ou de
pré-résonance, voir ce chapitre, II.1.3).
Les éléments
analysés doivent également rester stables sous l’effet d’un échauffement
local possible, conséquence directe du phénomène dont il a été question
dans le point précédent. Selon Merlin (1990), une focalisation
sur une surface de 1 µm² correspond à une densité d’énergie de 1 mégaWatt/cm².
La solution à ce problème passe par une réduction de l’intensité du laser
excitateur ou par une défocalisation du point d’impact.
La fluorescence,
phénomène fréquent dans de nombreuses méthodes spectroscopiques, l’est également
en MR. Elle constitue un inconvénient majeur dans l’étude de certains
produits organiques et plus rarement dans le cas de certains produits minéraux
(voir ci-dessous, II.1.3, la fluorescence).
Enfin, certaines phases régies par des
règles de symétries bien précises, ne peuvent donner de spectre Raman. Ces
phases, tels que NaCl ou tels que certains métaux (Au, Cu ...) sont identifiées
par les règles physiques de la théorie des groupes décrites ci-dessous.
La
MR est basée sur le phénomène des vibrations des liaisons entre les
atomes, et plus particulièrement sur la réponse de ces liaisons à une
excitation lumineuse. La MR fournit alors un spectre unique caractéristique à chaque
matériau.
Ces vibrations, identifiées par
les différents décalages de fréquence de la lumière visible, ultraviolette
ou infrarouge diffusée par l’échantillon, dépendent de la cristallochimie
du minéral et elles peuvent nous renseigner sur (Dhamelincourt et Bisson,
1977; Pinet et al., 1992) :
Ø la nature du cristal (à la fois sa structure et sa composition
chimique) ;
Ø l’état de cristallinité d’un minéral ;
Ø son degré d’ordre.
Le principe de l’identification
d'un spectre obtenu consiste alors à comparer l’empreinte spectrale d'un
matériau inconnu à celle d’échantillons « étalons », à condition
bien sûr, de disposer d'une base de données suffisamment étoffée (Smith & Edwards,
1998).
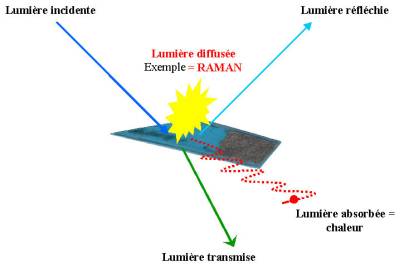
La lumière est réfléchie par la surface de
particules dont l’orientation aléatoire provoque une distribution de
l’intensité lumineuse dans toutes les directions de l’espace
(Bouguer, P., Traité sur la gradation de la lumière, Paris , 1760).
Le principe
physique de cette méthode repose sur les différentes manières dont la lumière
est renvoyée lorsqu’elle arrive sur la surface de l’objet (autrement dit,
une couche d’atomes).
Ø une partie de cette lumière est réfléchie par la
première couche d’atome ;
Ø une seconde est transmise dans les cristaux isotropes
ou anisotropes selon des règles d’optique ;
Ø une troisième est absorbée et transformée en chaleur,
Ø et
enfin une quatrième est diffusée dans toutes les directions de l’espace.
Pour toute
liaison entre les atomes d’une molécule ou d’un cristal vibrant à une fréquence nv, on peut
faire correspondre une valeur particulière de l’énergie de la molécule.
On représente ces valeurs par E0, E1, E2,
E3, etc. L’écart entre ces niveaux d’énergie est noté DE et on note :
DE = E1- E
0 =
h
nv = hc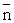 v
v
On préfère alors aux fréquences de vibrations
moléculaires de l’ordre de ~1012 ou 1013 Hertz,
une graduation de l’échelle horizontale en « nombre d’onde », fonction
reliée à la fréquence et à la longueur d’onde par une relation suivante :
avec h = constante de Planck, c = vitesse de la lumière, n =
fréquence,
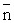
=
nombre d’onde et
l= longueur d’onde.
Ainsi, le nombre d’onde est l’inverse
de la longueur d’onde (cm) et s’exprime en cm-1. Précisons également
que le zéro de l’échelle des nombres d’ondes relatifs coïncide avec la radiation
excitatrice (autrement dit, c’est la bande Rayleigh -le rayonnement excitateur-
sur le spectre Raman qui sera à la position « 0 cm-1 » de
nombre d’onde relatif).
D’autre part,
grâce à la loi de distribution de Boltzmann, on peut calculer pour un système à l’équilibre
le nombre de molécules possédant à chaque instant les énergies E0,
E1, E2, E3, etc. On note ce nombre NE0, NE1, NE2, etc.,
il correspond alors aux populations des différents niveaux d’énergie. Le
rapport de ces nombres entre deux niveaux successifs s’exprime
par l’ équation d’Arrhénius :
k = constante de Boltzmann, T = température absolue.
Cela revient à dire que le niveau inférieur
est le plus peuplé et que sa population décroît exponentiellement, en fonction
de l’énergie du niveau considéré.
L’énergie transportée par un quantum
ou « photon » de lumière excitatrice est égale à hn0 . Une grande partie
de la lumière diffusée qui sort de l’échantillon a la même longueur d’onde que la lumière excitatrice : c’est la diffusion Rayleigh ou élastique.
Cependant,
une infime partie (10-9 à 10-12 fois) est
diffusée avec une longueur d’onde différente de la longueur d’onde excitatrice
(h
n0) ; ce sont les diffusions RAMAN et Brillouin
ou
encore inélastiques.
Remarque : L'effet Raman résulte de l'interaction
des ondes électromagnétiques avec les modes vibrationnels ou rotationnels
des liaisons des atomes d'une molécule, alors que l'effet Brillouin est
quant à lui basé sur l'interaction de ces ondes avec les modes translationels
des liaisons de ces atomes.
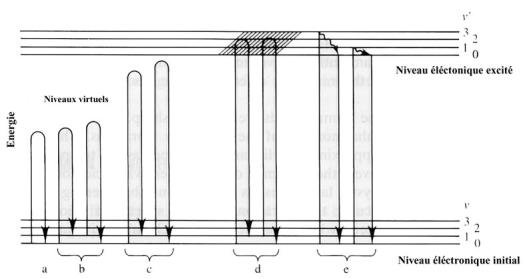
Ainsi, comme le montre la figure ci-dessous :
si l’électron,
excité par la longueur d’onde incidente -hn0- retombe dans le même niveau d’énergie de départ E0, (il réémet
alors des photons à la même longueur d’onde hn1= hn0), il s’agit de diffusion Rayleigh dont il a été question ci-dessus.
si l’électron,
excité par la longueur d’onde incidente retombe dans un niveau d’énergie
supérieur au niveau de départ (il émet alors une longueur d’onde plus courte
que la longueur d’onde incidente), on parle de diffusion Raman Stokes (E1<E0 et DE est négative).
Enfin, si l’électron, excité par la longueur
d’onde incidente retombe dans un niveau inférieur d’énergie, émettant une
longueur d’onde plus grande, on parle alors de diffusion Raman anti-Stokes (E1>E0 et DE est positive).
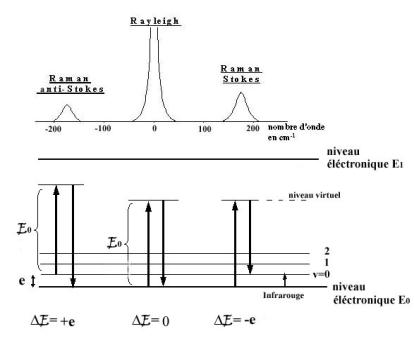
La probabilité d’observation de transitions
anti-Stokes, partant de niveaux moins peuplés, est plus faible que celle
qui est observée pour les transitions Stokes ; de plus, elle décroît
exponentiellement en fonction de l’énergie du niveau de départ. Cette constatation,
couplée à la symétrie qui existe entre les vibrations Stokes et anti-Stokes
par rapport aux vibrations Rayleigh, conduit à choisir les vibrations Stokes
pour l’étude Raman classique. Même si la grande majorité des chercheurs
adoptent les vibrations Stokes avec un signe positif de nombre d’onde (comme
sur le schéma ci-dessus), il peut arriver que certaines présentations inversent
les deux vibrations, de sorte que les vibrations Stokes apparaissent en dessous
de la bande excitatrice (ex : Ferraro & Nakamoto, 1994).

En général,
la plupart des spectres Raman des minéraux présentent des bandes situées
entre 100 cm-1 (et ceci afin que le signal Raman ne soit pas
perturbé par la bande Rayleigh très intense) et 1600 cm-1, ainsi
que des bandes situées dans des zones spécifiques à certaines vibrations
de la molécule (ex : 3450-3750 cm-1 pour les liaisons O—H).
(Encycl.Univ., 1995)
Une molécule à N atomes peut être traitée
comme un système mécanique à 3 N degrés de liberté, parmi lesquels on
compte trois translations d’ensemble, trois rotations (ou deux rotations
seulement pour une molécule linéaire). Les 3 N - 6 (ou 3 N - 5) mouvements restants
rendent compte des déformations internes à la molécule, qui correspondent
aux vibrations moléculaires.
Le traitement classique des vibrations
d’une molécule polyatomique consiste à résoudre un système d’équations de
Lagrange, dans lesquelles interviennent les énergies cinétique et potentielle.
Dans l’hypothèse harmonique, le mouvement peut se résoudre en un ensemble
de « modes normaux », auxquels sont associées des « coordonnées
normales ». Pour chacun d’eux, tous les atomes vibrent en phase à la
même fréquence, mais avec des amplitudes différentes. L’analyse des 3 N - 6 vibrations d’une molécule à grand
nombre d’atomes ne peut être menée qu’en mettant en œuvre les moyens de calcul
puissants disponibles de nos jours. Cependant, une analyse simplifiée, ne
mettant en jeu que les propriétés de symétrie d’un édifice polyatomique,
peut suffire si l’on se contente de dénombrer les modes vibrationnels et
de déterminer leur activité en absorption infrarouge ou en diffusion Raman.
Cette activité, qui détermine la possibilité d’observation
d’un mode vibrationnel donné, soit par absorption, soit par diffusion lors
de l’interaction avec un rayonnement, peut être corrélée à des « règles
de sélection ». Ces dernières gouvernent les possibilités de couplage
de l’onde électromagnétique avec le mouvement moléculaire, lorsque celui-ci
s’accompagne d’une variation des propriétés liées à la polarisation électrique
de la molécule : moment dipolaire
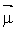
et
polarisabilité moléculaire
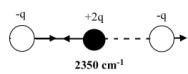
a.Moment dipolaire : Le moment dipolaire
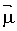
est une
propriété vectorielle. Lorsque le barycentre des charges positives (noyaux
atomiques) ne coïncide pas avec celui des charges négatives (électrons),
la molécule est assimilable à un dipôle électrique formé de deux charges
ponctuelles
+ q et -q situées à une distance
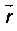
:
Polarisabilité moléculaire : La polarisabilité moléculaire
traduit, quant à elle, la faculté de déformation du nuage de charges électriques
sous l’influence d’un champ électrique uniforme, et se définit par :
où
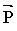
est le moment électrique induit et
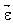
le vecteur champ électrique.
La direction du moment induit ne coïncide pas, en général, avec celle du
champ, et la valeur de
a varie
selon l’orientation de la molécule.
En fait, a n’est pas, comme pourrait le laisser penser
la formule ci-dessus, un simple coefficient de proportionnalité, mais correspond à un
tenseur de rang 2 représenté par une matrice d’ordre 3 symétrique. Chaque
composante du champ E0 peut induire une déformation du nuage électronique,
donc un moment induit dans les trois directions de l’espace (Felidj et
al., 1998) :
Px axx axy axz Ex
Py = ayx ayy ayz Ey
Pz azx azy azz Ez
On pourra
trouver, en partie, les développements du « traitement classique » de
l’effet Raman dans la publication de Felidj et al., (1998).
Les règles de sélection peuvent se résumer
de la manière suivante :
Une vibration de coordonnée normale Q
peut donner lieu à une absorption Infrarouge, si elle
modifie
l’une des composantes du
moment dipolaire 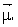
, c’est-à-dire
si la dérivée
(bmi /bQ)0 n’est pas nulle au voisinage de la position
d’équilibre.
Une vibration de coordonnée normale Q
est active en diffusion Raman, si elle fait varier
une des composantes aij de la polarisabilité, c’est-à-dire
si la dérivée (baij /bQ)0 n’est pas nulle.
Si les variations du moment électrique
et de la polarisabilité paraissent intuitivement accessibles pour des molécules
di- ou triatomiques, il n’en est pas de même pour des édifices polyatomiques à grand
nombre d’atomes.
Une méthode très élégante permet de tourner
cette difficulté, en se fondant uniquement sur les propriétés de symétrie
des molécules, et en faisant appel à des résultats connus de la théorie
des groupes. Toutes les propriétés utiles à la prévision des spectres
vibrationnels sont contenues dans la table de caractères du groupe.
Les 3 N mouvements de la molécule, translations, rotations et modes
normaux de vibration, se rangent dans cette table en fonction de leurs propriétés
de symétrie, ainsi que les 3 composantes du vecteur moment dipolaire
et les 6 du tenseur de polarisabilité.
L’analyse vibrationnelle, c’est-à-dire
le dénombrement et l’activité des modes de vibration en Raman ou Infrarouge,
s’effectue à partir de ces tables par un calcul très simple où intervient
le nombre d’atomes inchangés au cours d’une opération de symétrie.
Voici certaines règles simples qui concernent
les modes actifs et inactifs en Raman et IR :
Ø Certains
modes de vibration peuvent être inactifs à la fois en diffusion Raman et
en absorption Infrarouge.
Ø Si
la molécule possède un centre de symétrie, il n’existe aucune vibration commune
aux spectres Raman et infrarouge : celles qui sont symétriques par rapport à ce
centre (g, de l’allemand gerade ) sont actives en Raman
et inactives en infrarouge. Les vibrations antisymétriques par rapport à ce
centre (u,
de l’allemand ungerade) sont au contraire actives dans le domaine de l’infrarouge
et inactives en Raman (règle de mutuelle exclusion). La présence simultanée
de certains modes, à la fois dans les spectres Raman et Infrarouge, indique
de façon certaine l’absence de centre de symétrie.
Ø La
présence d’au moins un axe de symétrie d’ordre supérieur à deux se traduit
par l’apparition de modes dégénérés, donc confondus à une même fréquence.
Le nombre apparent de bandes ou de raies peut ainsi devenir inférieur aux 3N-6 prévus. C’est le cas classique,
par exemple, des édifices tétraédriques, comme CCl4, qui ne
présentent
que 4 bandes au lieu de 9.
Ø Les
vibrations symétriques sont toutes actives en Raman et se traduisent par
des raies intenses et polarisées (par exemple, pulsation symétrique du benzène
et des cycles aromatiques). Les vibrations non totalement symétriques ou
dégénérées donnent des raies dépolarisées.
En plus des modes fondamentaux prévus
dans l’hypothèse harmonique, des bandes correspondant à des «harmoniques » ou à des « combinaisons » de
modes apparaissent aussi dans les spectres expérimentaux. Ces bandes s’interprètent
par des transitions entre niveaux non consécutifs dans le diagramme énergétique.
Leur intensité est souvent beaucoup plus faible dans le spectre Raman que
dans l’Infrarouge.
Exemples partiels des molécules de N2 et
de CO2.
Lorsque la radiation excitatrice est
proche d’une bande d’absorption électronique de la molécule, on observe une
exaltation de l’intensité de certaines raies du spectre Raman. Cette exaltation
s’interprète par un effet de résonance. En théorie, il faut distinguer la
résonance exacte, dans laquelle l’excitation coïncide avec le maximum d’absorption,
de la pré-résonance, plus fréquemment observée, puisqu’il suffit de s’approcher
des bandes électroniques du chromophore moléculaire (c’est-à-dire du groupe
d’atomes qui, dans la molécule, est à l’origine de sa « couleur »)
pour que l’exaltation se produise.
L’accroissement de l’intensité, qui peut
atteindre des valeurs très élevées (106 dans des cas favorables),
résulte d’un couplage des transitions vibrationnelles et électroniques ;
les raies exaltées correspondent à la structure vibrationnelle de la bande
d’absorption électronique mise en jeu. Le phénomène peut cependant se compliquer
car l’interaction vibrationnelle peut concerner non plus un seul niveau électronique
excité, mais aussi le couplage vibronique de deux états électroniques excités.
L’état de polarisation des spectres de résonance peut être très différent
de ce qui est observé en diffusion Raman ordinaire, si bien que les mesures
de polarisation apportent des données très utiles à l’interprétation des
spectres.
En pratique, les effets de résonance
sont mis à profit, grâce à un choix judicieux de la radiation excitatrice,
pour exalter sélectivement le spectre de vibration de certaines molécules
(dans un mélange complexe ou une solution diluée), d’un groupement d’atomes
impliqué dans un chromophore porté par une macromolécule.
Cet effet sera particulièrement mis à profit
dans les différentes études menées dans ce mémoire, la plupart des espèces
minérales analysées étant fortement colorées et donc fortement susceptibles
d’entrer en résonance avec la radiation excitatrice utilisée.
S’il arrive qu'il faille décomposer le spectre Raman en détail pour identifier
l’espèce étudiée, souvent, il suffit d'une simple comparaison avec un autre
spectre de référence pour identifier l’espèce en question.
Les propriétés des bandes Raman sont très différentes selon l'état physique
et le degré de cristallinité du matériau étudié. Ainsi, le verre amorphe
donnera des bandes larges (« des bosses ») alors que les minéraux
bien cristallisés donneront de très fines bandes. L'épaisseur de ces bandes
est principalement due à l'anharmonicitée des vibrations moléculaires. En
outre, que le matériau soit gazeux, liquide ou solide, l'allure du spectre
dépendra des conditions d'analyse (température, pression ...). Dans l’état
cristallin, les effets thermodynamiques (comme une élévation de température)
viennent s'ajouter à des effets de structure (impuretés, sites vacants, imperfections
de la maille cristalline, états amorphes ...) et influent de la sorte sur
la largeur des bandes Raman.
On ne traite dans ce mémoire que de matériaux
se trouvant dans un état solide/cristallin, amorphe ou vitreux ; nous
ne développons donc pas les caractéristiques propres à l’état liquide et
gazeux, points déjà largement détaillés dans d’autres ouvrages tels que Ferraro & Nakamoto
(1994) ou Turrell & Corset (1996).
Enfin, l’opération
consistant à lisser les spectres Raman « bruts » n’est
pas acceptée par tous les spécialistes ; en effet, le bruit de fond
observable sur un spectre est difficilement éliminable sans nuire aux pics
réellement caractéristiques de l’échantillon. Dans notre cas, un lissage
partiel et une légère correction de la ligne de base ont été effectués
sur les spectres qui en présentaient vraiment la nécessité.
Un point délicat de la Microscopie Raman,
lié à l’intensité des signaux, doit encore être soulevé :
il s’agit de l’orientation des cristaux.
En effet,
selon cette orientation, l’intensité (et non la longueur d’onde)
peut varier (on retrouve de la même façon ce problème avec d’autres méthodes
telles que la diffraction-X). Il est donc souhaitable de mesurer les cristaux
sous un minimum de deux orientations (axe cristallographique, plan de clivage,
axe optique, etc.) (Smith, 1996).
Un autre point primordial lié à l’intensité absolue
des bandes Raman est l’angle solide de collecte de la lumière diffusée, autrement
dit, l’intensité collectée (Ω), fonction de l’ouverture numérique de
l’objectif (ON) (Univ. du Maine, 2000) ;
Cette variable
est notée : Ω = 2p[1- cos(a)]
L’ouverture numérique et l’angle de collecte
(a) sont généralement fournis par
le fabricant. Le tableau suivant montre la surface analysée en fonction
de la valeur de a (selon Univ. du Maine, 2000).
La fluorescence représente un des problèmes majeurs de la MR : en
effet, une fois excitée par le laser, cette fluorescence masque le signal
Raman du produit. Elle serait, d’après Wopenka (1990), plus importante
dans les minéraux contenant du fer, mais il est vrai que la fluorescence
est en réalité un phénomène beaucoup plus courant sur les matériaux organiques
(Pasteris, 1988).
Le problème de la fluorescence est particulièrement prononcé dans ce
domaine précis où les matières organiques « cohabitent » souvent
avec les matières minérales, comme c’est le cas pour de nombreux pigments.
Il arrive donc fréquemment que le signal Raman soit quelque peu, voire
totalement, masqué par la fluorescence.
Ce fait trouve une explication
dans la durée respective de ces deux phénomènes (Turell & Corset,
1996). Dans le cas de l’effet Raman, la durée de désexcitation
des électrons
est inférieure à quelques picosecondes (10-12s) (voir
figure 5), alors que dans le cas
de la fluorescence ou de la phosphorescence, l’effet atteint en moyenne
les 10-9 secondes,
temps suffisamment long pour masquer totalement ou en partie l’effet
Raman.
Il existe certaines règles ou astuces
qui permettent de réduire ou d’éliminer la fluorescence d’un échantillon.
La plus simple, mais quelquefois négligée, quand il s’agit d’un échantillon
hétérogène (ex : des prélèvement de pigments de peinture) est de choisir,
grâce au microscope, une autre zone d’analyse ne présentant pas de défauts
de fluorescence. Il est en effet courant que dans un même échantillon,
selon la taille des cristaux par exemple, les différents grains ne « fluorescent » pas
de la même manière.
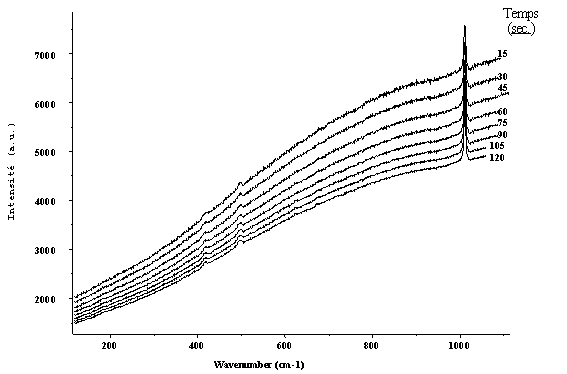
Dans certains cas, la fluorescence
peut même être réduite en prolongeant l'exposition sous le laser à très
faible puissance, afin de ne pas endommager l'échantillon, l’intensité des
bandes Raman restant constante. Cette expérience a été réalisée sur certains échantillons
particulièrement fluorescents (figure ci-dessous).
Une autre solution est d'utiliser
une source lumineuse de plus faible énergie (de type « proche Infra-rouge » (NIR) à 1064
nm) qui ne pourra pas provoquer les transitions d'électrons responsables
de la fluorescence. Cependant, pour compenser la faible réponse du signal
(18 fois moindre), il est alors nécessaire d'utiliser une méthode de transformation
de Fourier (Chase, 1987).
De nombreuses autres méthodes,
plus ou moins efficaces, tentent de combattre la fluorescence. On peut
citer un dernier exemple (le plus efficace, mais aussi le plus onéreux)
qui consiste à déterminer le temps nécessaire à l'obtention d'une réponse
Raman, souvent instantanée, par rapport au temps nécessaire à une réponse
de la fluorescence électronique, souvent beaucoup plus long (Kamogawa,
1988 ; Sharma, 1989). Il ne reste plus par la suite qu’à fixer la
durée de l’excitation par le laser (laser à impulsion -de l’ordre de la
nanoseconde-) ainsi que celle de l’acquisition correspondante.
Le rayonnement émis par les lasers à gaz est un autre type de
problème auquel nous avons dû faire face. Ce rayonnement, qui n’est pas
strictement monochromatique, provoque l’émission de raies additionnelles
dites ‘plasma’ qui peuvent fausser les résultats si elles ne sont
pas éliminées. Une technique couramment usitée consiste à utiliser un filtre
‘interférentiel’ spécifique à chaque longueur d’onde excitatrice
qui permet de filtrer ces raies plasma avant que le laser ne pénètre
dans le microscope.
Il est cependant possible que certaines
raies, notamment vers les petites longueurs d’ondes, (ex : l’intense
raie à ~117 cm-1) réussissent à passer à travers le filtre interférentiel
et se retrouvent dans le spectre Raman du produit analysé.
D’autres phénomènes peuvent venir
perturber les analyses ; il s’agit des différents éléments environnants
qui peuvent, soit se superposer à l’excitation du laser (par exemple la
lumière de la salle), soit encore, interférer avec le laser (ex :
les couches de protection internes des objectifs du microscope -de type Olympus en
l’occurrence-). Ce dernier phénomène se produit assez souvent, de sorte
que toutes les bandes caractéristiques de cette couche ont été clairement
identifiées afin qu’aucune n’interfère avec les bandes Raman réelles de
l’échantillon analysé. Ces bandes sont principalement observées lors de
l’utilisation d’intensités élevées comme c’est le cas lors de l’analyse
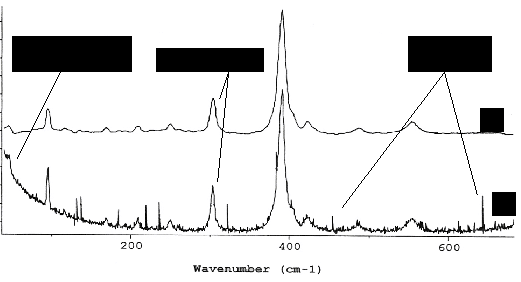
des verres (intensité à la source ~ 1W) (voir spectre Raman 4).
Spectre Raman « brut » de la couche interne de l’objectif Olympus -BGPA04-
Bandes à 844, 895 et 921 cm-1 avec des intensités relatives
toujours respectées.
(Encycl.Univ., 1995).
La fenêtre spectrale la plus utilisée
pour l’analyse de l’effet Raman se situe entre les absorptions électroniques
(ultraviolet) et vibrationnelles (infrarouge). C’est précisément dans cette
gamme de longueur d’onde que l’on dispose le plus facilement des différents
matériaux optiques et électroniques de haute performance.
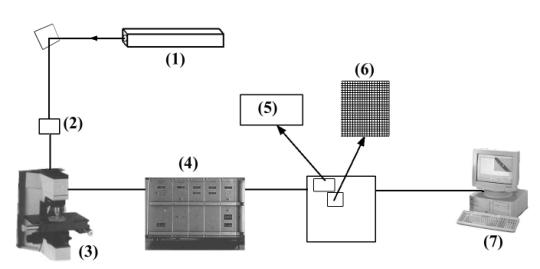
La figure 6 résume schématiquement le
fonctionnement d’un spectromètre Raman. En voici énumérés les principaux
organes :
La source laser, qui délivre, dans un
faisceau de très faible divergence, une radiation monochromatique polarisée.
Un filtrage soigné débarrasse ce faisceau des raies du plasma ou du fond
continu de lumière incohérente provenant du dispositif de pompage optique
du milieu actif.
Des optiques de transfert (miroirs, etc.)
chargées de diriger la lumière excitatrice vers le microscope.
Le microscope, où l’on retrouve les différents
objectifs chargés de l’illumination optimale de l’échantillon et de la collection
la plus efficace possible de la lumière diffusée par effet Raman, ainsi que
de son transfert vers l’entrée du spectromètre proprement dit. L’objet à analyser
est posé sur le porte-échantillon du microscope.
Le spectromètre, qui effectue l’analyse
spectrale du rayonnement et qui comporte généralement plusieurs étages de
disperseurs à réseaux, de 2 à 4 disposés en cascade, de manière à accroître
la pureté spectrale du système d’analyse.
Le spectromètre comporte soit un prémonochromateur,
soit un filtre holographique qui élimine très efficacement les radiations
réfléchies ou diffusées à la longueur d’onde du laser, suivi d’un spectrographe
dispersif. Un autre type d’instrument, plus spécialement adapté à l’excitation
dans le proche infrarouge à 1,06 µm, afin d’éviter la fluorescence,
réalise l’analyse spectrale grâce à un interféromètre à transformée de
Fourier couplé à un micro-ordinateur. L’avantage le plus évident du système
multicanal réside dans la simultanéité des mesures de toutes les composantes
spectrales. Toutes les informations, perdues au cours de l’opération de balayage
dans un spectromètre monocanal, sont conservées et mises en mémoire grâce
au système multicanal, ce qui améliore le rapport signal/bruit et permet
de détecter des raies qui, antérieurement, n’émergeaient pas du bruit de
fond. La simultanéité des mesures permet de s’affranchir des variations d’intensité de
la source de lumière et même d’enregistrer des spectres Raman excités par
des lasers en impulsions de durée très brève (de 10 nanosecondes à quelques
dizaines de picosecondes).
Le photomultiplicateur qui traduit les
photons en signal électrique.
Le détecteur photoélectrique : deux
dispositions sont adoptées selon le type de détecteur photoélectrique employé.
Dans les spectromètres du type « monocanal», utilisant un photodétecteur
unique, l’analyse des différents éléments du spectre est effectuée séquentiellement,
par rotation des axes des réseaux, de manière à faire défiler les différentes
radiations focalisées dans le plan d’une fente qui isole une bande passante étroite.
Dans une génération plus performante d’instruments appelés « spectromètres
multicanaux », le détecteur photoélectrique est constitué d’une mosaïque
du type CCD (charge coupled device) refroidie à basse température.
La dernière partie de l’instrument assure
la mesure et le traitement des signaux électriques issus des détecteurs.
Le développement des techniques numériques de traitement et la puissance
des systèmes micro-informatiques disponibles ont permis d’accroître la qualité des
mesures spectroscopiques et d’assurer, en même temps, la gestion des différentes
fonctions du spectromètre.
Afin de ne
pas provoquer d’échauffement local et de ne pas brûler les échantillons
photosensibles, il est de mise de commencer l’analyse avec une intensité lumineuse
faible et de l'augmenter au fur et à mesure. On peut également maintenir
une intensité faible et prolonger la durée d'analyse.
Depuis peu, l’intérêt porté à la
spectrométrie Raman par les archéologues, les industriels ou les scientifiques
de tous bords a favorisé le développement de microsondes « mobiles » (MRM
-Smith, 1999-) ou « transportables ».
Dans le premier cas, l’appareillage nécessite
d’être porté par trois ou quatre personnes, dans le second, l’appareillage
est plus miniaturisé et peut être porté par une seule personne. Il est même
prévu (à moyen terme, ~2008) d’envoyer sur la planète Mars une Microsonde
Raman de la taille d’une cannette de soda afin d’effectuer des analyses extraterrestres
de la planète en question (exemple : Wang, 1996, 1998).
Pour un gaz ou un liquide dit « optiquement vide » (c’est-à-dire
dont les poussières ou les particules diffusantes ont été éliminées par
des filtrations répétées) le rapport des intensités Stokes / Rayleigh est
de l’ordre de 10-3.